Detecting Positive Selection at Single Sites (Suzuki and Gojobori 1999)
Selective forces operating at the amino acid sequence level have been detected mainly by comparing the nonsynonymous substitutions per site with that of synonymous substitutions per site with that of synonymous substitutions per site. Generally speaking, the excess number of synonymous substitutions was considered to be the result of negative selection, whereas that of nonsynonymous substitutions was attributed to positive selection.
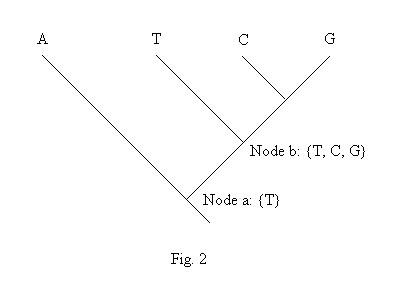
Suzuki and Gojobori (1999) proposed a method to detecting positive selection at single amino acid sites, assuming that the synonymous change was almost neutral. The method can not be used when your sequences are non-coding sequences Most algorithms used in the package are identical with those in Suzuki and Gojobori (1999). The package does not implement the proposed rule, that is when the number of combinations for possible ancestral codons over all nodes exceeded 10,000, that site will be excluded. Moreover, we changed the genetic distance for Neighbor-Joining (NJ) tree, and "revised" Hartigan's method. The reason for the latter is presented here.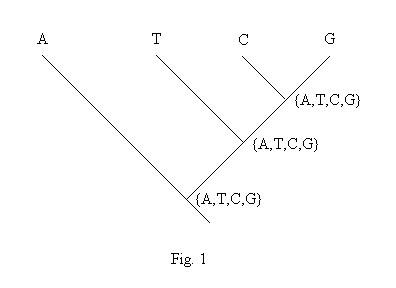
- st -- the average number of synonymous sites throughout the phylogenetic tree
- nt -- the average number of nonsynonymous sites throughout the phylogenetic tree
- sc -- the total number of synonymous mutations throughout the phylogenetic tree
- nc -- the total number of nonsynonymous mutations throughout the phylogenetic tree
To detect the negative selection, given the fact of that, mut = 4 (sc) + 1 (nc), the probability of that nc is equal to or more than 1 is calculated by binomial distribution. It will be labeled by blue color and asterisk when the probability is less than 5%.