EpiDISH (Epigenetic Dissection of Intra Sample Heterogeneity)
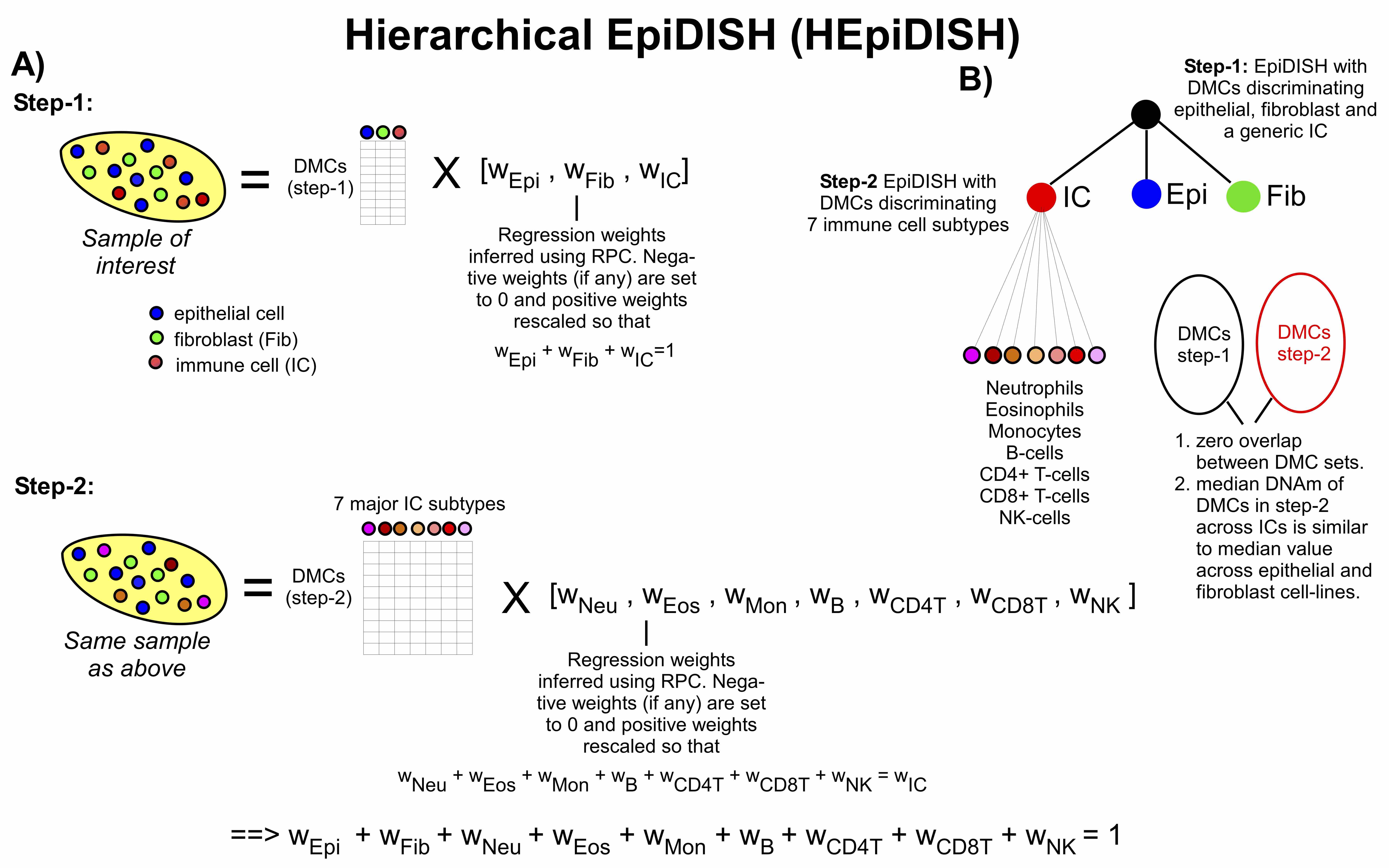
The EpiDISH package provides tools to infer the proportions of a priori known cell subtypes present in a sample representing a mixture of such cell-types. Inference proceeds via one of 3 methods (Robust Partial Correlations-RPC(Teschendorff et al. 2017), Cibersort (CBS)(Newman et al. 2015), Constrained Projection (CP)(Houseman et al. 2012)), as determined by user. Besides, we also provide a method - CellDMC that allows the identification of differentially methylated cell-types and their directionality of change. For now, the package contains 4 references, including two blood subtypes reference, as well as one reference with epithelial cells, fibroblasts, and total immune cells, and one reference with epithelial cells, fibroblasts, fat cells, and total immune cells, described in (Teschendorff et al. 2017) and (Zheng, Webster, et al. 2018).
Maintainer: Shijie Charles Zheng
Citation
Teschendorff, Andrew E, Charles E Breeze, Shijie C Zheng, and Stephan Beck. 2017. “A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies.” BMC Bioinformatics 18 (1):105.
Teschendorff, Andrew E, and Shijie C Zheng. 2017. “Cell-type deconvolution in epigenome-wide association studies: a review and recommendations.” Epigenomics 9 (5):757–68.
Zheng, Shijie C, Charles E Breeze, Stephan Beck, and Andrew E Teschendorff. 2018. “Identification of differentially methylated cell-types in Epigenome-Wide Association Studies.” Nature Methods 15 (12):1059–66.
Zheng, Shijie C, Amy P Webster, Danyue Dong, Andy Feber, David G Graham, Roisin Sullivan, Sarah Jevons, et al. 2018. “A novel cell-type deconvolution algorithm reveals substantial contamination by immune cells in saliva, buccal and cervix.” Epigenomics 10 (7):925–40.
Installation
To install this package, start R (version "3.6") and enter:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("EpiDISH", version = "3.9")Documentation
To view documentation for the version of this package installed in your system, start R and enter:
browseVignettes("EpiDISH")HTML: EpiDISH Vignette
SEPIRA (Systems EPigenomics Inference of Regulatory Activity)
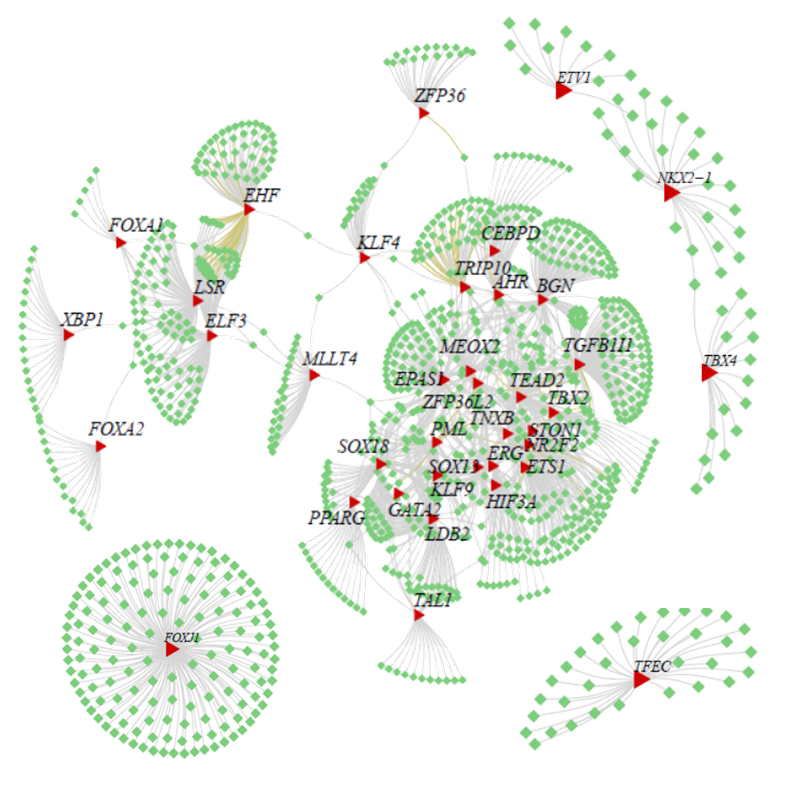
SEPIRA is a novel algorithm which estimates transcription factor activity in any given sample from its genome-wide mRNA expression or DNA methylation profile(Chen Y, Widschwendter M, and Teschendorff AE 2017). It encompasses two main steps: 1.Construction of a tissue-specific transcription factor regulatory network, consisting of transcription factors that are more highly expressed in the user-specified tissue type (the ‘tissue type of interest’) compared to other tissue types, plus an associated set of high-confidence downstream targets. 2.Estimation of transcription factor activity in this network, in any given dataset consisting of gene expression or promoter DNA methylation profiles.
Maintainer: Yuting Chen
Installation
To install this package, start R (version "3.5") and enter:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("SEPIRA", version = "3.8"Documentation
To view documentation for the version of this package installed in your system, start R and enter:
browseVignettes("SEPIRA")HTML: Introduction to SEPIRA
ChAMP (The Chip Analysis Methylation Pipeline)
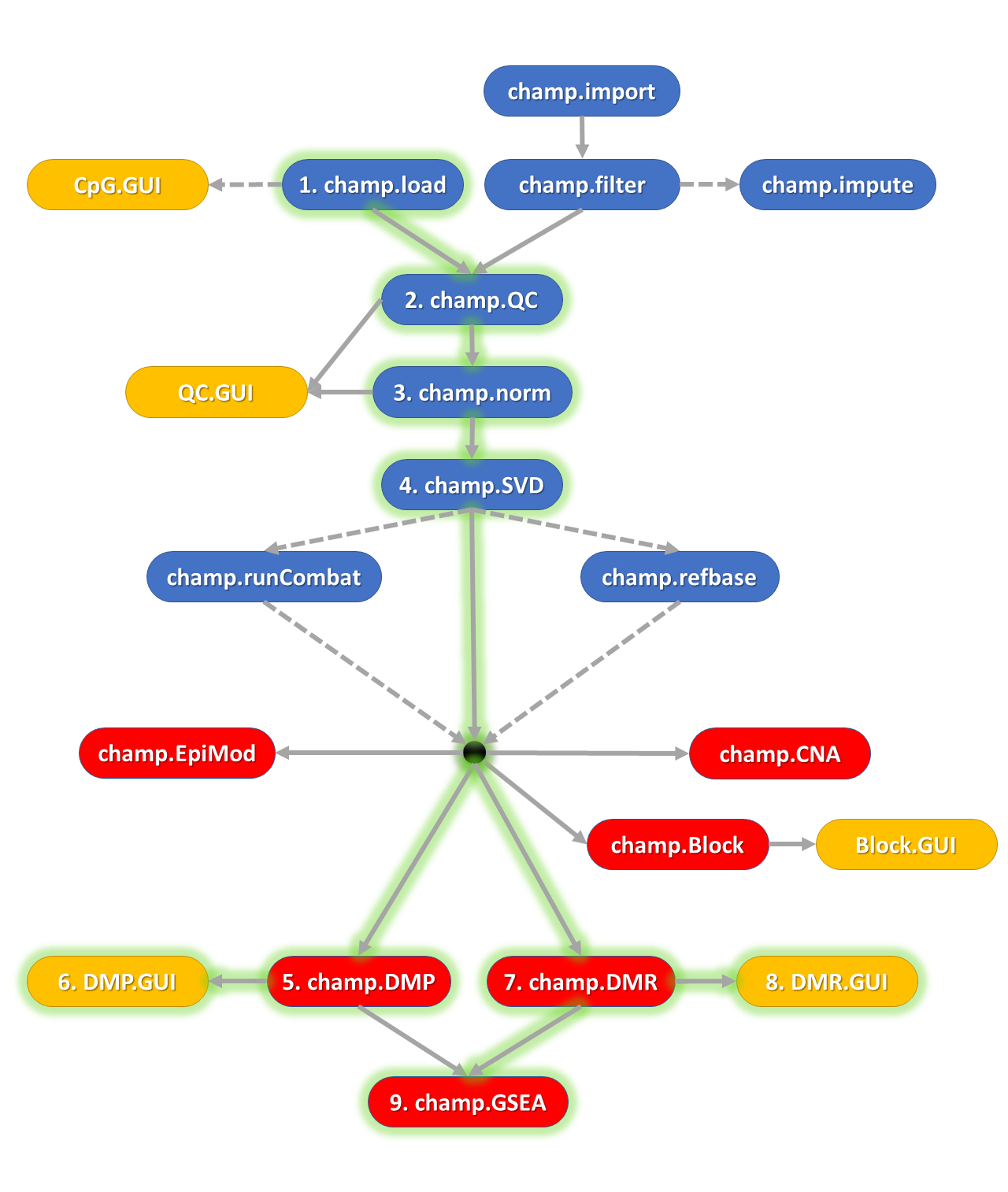
The ChAMP package is designed for the analysis of Illumina Methylation beadarray data (EPIC and 450k) and provides a pipeline that integrates currently available 450k and EPIC analysis methods. The new version of ChAMP, extends and improves this analysis pipeline, adding novel and enhanced functionalities, including detection of differentially methylated genomic blocks (DMB), Gene Set Enrichment Analysis (GSEA), a variety of methods for correcting cell-type heterogeneity and detection of differentially methylated gene modules. Notably, the new package provides a series of web-based graphical user interfaces (GUIs), which facilitate analyses and enhance user-experience.
Maintainer: Yuan Tian
Citation (from within R, enter citation("ChAMP")):
Yuan Tian, Tiffany J Morris, Amy P Webster, Zhen Yang, Stephan Beck, Andrew Feber, Andrew E Teschendorff; ChAMP: Updated Methylation Analysis Pipeline for Illumina BeadChips, Bioinformatics, , btx513, https://doi.org/10.1093/bioinformatics/btx513
Installation
To install this package, start R and enter:
source("http://bioconductor.org/biocLite.R")
biocLite("ChAMP")Documentation
To view documentation for the version of this package installed in your system, start R and enter:
browseVignettes("ChAMP")HTML: ChAMP Tutorial
FEM (Functional Epigenetic Modules) algorithm
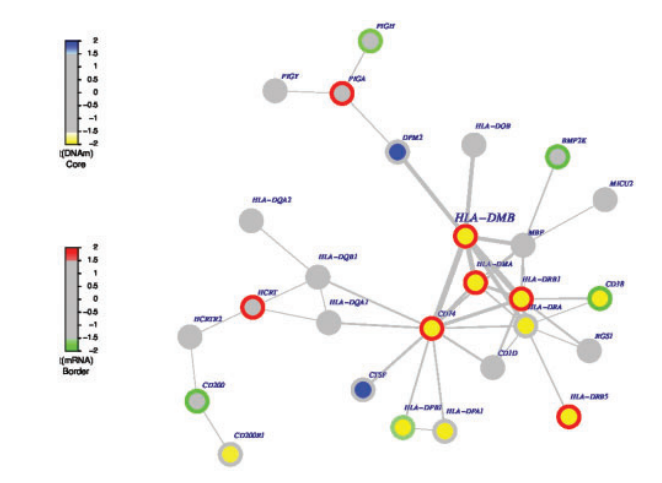
FEM can dentify interactome hotspots of differential promoter methylation and differential ex-pression, where an inverse association between promoter methylation and gene expression is assumed.A R package to identify interactome hotspots of differential promoter methylation and differential expression, where an inverse association between promoter methylation and gene expression is assumed1.
Installation
To install this package, start R and enter:
source("http://bioconductor.org/biocLite.R")
biocLite("FEM")Documentation
To view documentation for the version of this package installed in your system, start R and enter:
browseVignettes("FEM")PDF : A R package to identify interactome hotspots of differential promoter methylation and differential expression, where an inverse association between promoter methylation and gene expression is assumed1.
PDF: Reference Manual
BMIQ (Beta MIxture Quantile dilation) algorithm
A novel model-based intra-array normalisation strategy for 450k data, called BMIQ (Beta MIxture Quantile dilation), to adjust the beta-values of type2 design probes into a statistical distribution characteristic of type1 probes. BMIQ will be useful as a preprocessing step for any study using the Illumina Infinium 450k platform.
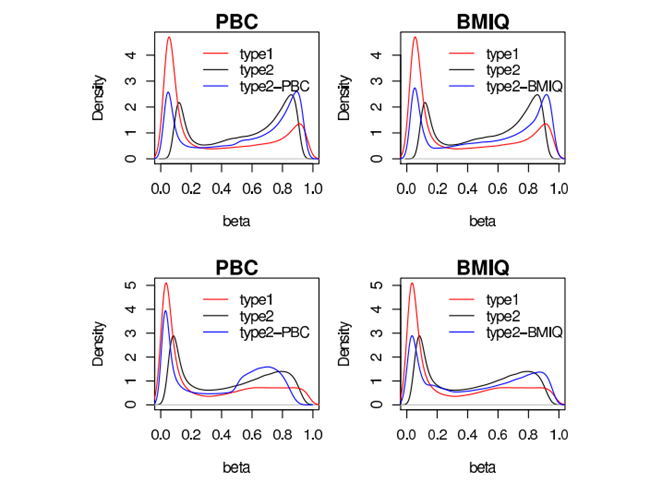
BMIQ
R Script for BMIQ This is the script of BMIQ
R Script for DoBMIQ This is a script to run BMIQ on a set of Methylation Data.
To run BMIQ code, after placing all stuffs(data, probe information, annotation) in one folder, you can use following R code to calculate it out:
source("DoBMIQ.R")Or you can find BMIQ function in ChAMP Package, which is developed by Tiffany Morris
Installation To install ChAMP package, start R and enter:
source("http://bioconductor.org/biocLite.R")
biocLite("ChAMP")ISVA (Independent Surrogate Variable Analysis) algorithm
We present a modified SVA, called Independent Surrogate Variable Analysis (ISVA), to identify features correlating with a phenotype of interest in the presence of potential confounding factors. ISVA should be useful as a feature selection tool in studies that are subject to confounding.
isva: Independent Surrogate Variable Analysis
Independent Surrogate Variable Analysis is an algorithm for feature selection in the presence of potential confounding factors.
Version: 1.8
Depends: qvalue, fastICA
Published: 2013-11-04
Maintainer: Andrew Teschendorff
DART (Denoising Algorithm using Relevance network Topology) algorithm
Denoising Algorithm based on Relevance network Topology (DART) is an algorithm designed to evaluate the consistency of prior information molecular signatures (e.g in-vitro perturbation expression signatures) in independent molecular data (e.g gene expression data sets). If consistent, a pruning network strategy is then used to infer the activation status of the molecular signature in individual samples.
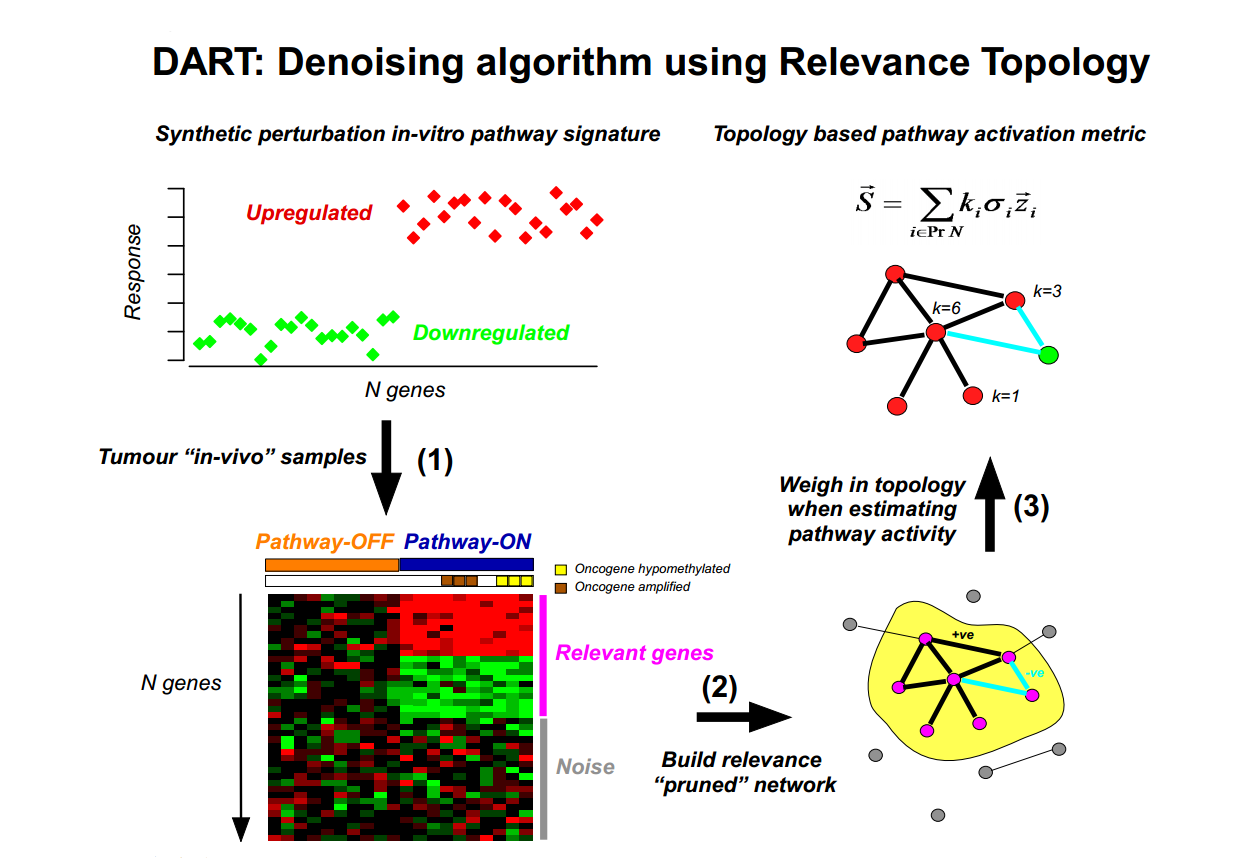
Maintainer: Katherine Lawler
Citation (from within R, enter citation("DART")):
Y J, K L, GS P, A P and al. e (2011). “DART: Denoising Algorithm based on Relevance network Topology improves molecular pathway activity inference.” BMC Bioinformatics, 12, pp. 403.
Installation
To install this package, start R and enter:
source("http://bioconductor.org/biocLite.R")
biocLite("DART")Documentation
To view documentation for the version of this package installed in your system, start R and enter:
browseVignettes("DART")PDF: DART Tutorial
PDF: Reference Manua
Signalling Entropy
we will describ e one approach which do es it at the level of individual samples, in which case the gene expression data must be p ositively valued (e.g. the expression data must derive from Affy, Illumina or RNA-Seq count data) and the mass action principle is used to construct the sto chastic matrix over the network..
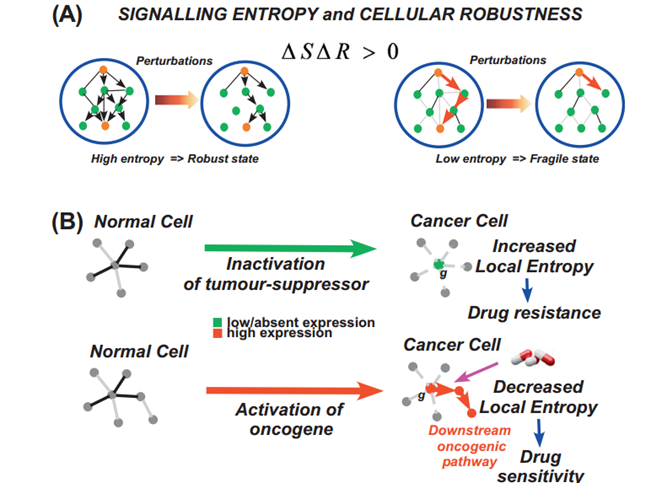
DESCRIPTION OF FILES:
1) dataSCM2.Rd: R data object file containing gene expression data matrix, avdataSCM2.m, from the stem cell matrix 2 compendium. Matrix is of size ~17000 genes X 191 samples. The type of cell (i.e hESC=1, iPSC=2 or somatic differentiated=3) is indicated in the phenoSCM2.lv[[1]] entry. Rownames of expression data matrix annotated to Entrez gene IDs.
2) hprdAsigH-13Jun12.Rd: R data object file with a model of PPI network specified by the adjacency matrix hprdAsigH.m. Rownames annotated to Entrez gene IDs.
3) sr.Rd: an auxiliary R object file needed to run the vignette.
4) DoIntegPIN.R: R-function to perform integration of a PPI network with a gene expression data matrix, extracting the maximally connected subnetwork.
5) CompSR.R: R-function to compute the entropy rate plus auxiliary functions.
6) CompSRana.R: R-function to compute the entropy rate analytically assuming detgailed balance (this will speed up the computation).
7) sigent.pdf: vignette/manual pdf.